¿Qué es la enfermedad de Fabry?
La enfermedad de Fabry, también llamada enfermedad de Anderson-Fabry, es una rara enfermedad hereditaria, transmitida de forma recesiva en el cromosoma X, el cromosoma sexual femenino.
Se estima que solo 1 de cada 20.000 a 40.000 hombres tiene la mutación responsable de los síntomas de la enfermedad.
Los pacientes con la enfermedad de Fabry tienen una deficiencia de una enzima llamada alfa-galactosidasa A, que provoca problemas renales, cardíacos y del sistema nervioso.
En este artículo explicaremos la enfermedad de Fabry, hablando de su transmisión genética, sus causas, síntomas y opciones de tratamiento.
Genética
Todos los humanos tienen dos cromosomas sexuales. Los hombres tienen un cromosoma sexual Y y un cromosoma sexual X (los hombres son XY). Las mujeres, en cambio, tienen dos cromosomas sexuales X y ningún cromosoma Y (las mujeres son XX).
Cada uno de los cromosomas sexuales que poseemos ha sido transmitido por uno de los padres. Por ejemplo, cuando una pareja tiene un hijo varón, esto significa que el niño recibió el cromosoma Y de su padre y el cromosoma X de su madre, formando así el par XY. Cuando la pareja tiene una niña, esta recibe un cromosoma X de su padre y un cromosoma X de su madre, formando así el par XX.
Por lo tanto, como la madre solo puede transmitir el cromosoma X a sus hijos, quien «decide» el sexo del niño es siempre el cromosoma paterno. Si el padre transmite la Y, será un niño; si transmite la X, será una niña.
La enfermedad de Fabry es una enfermedad transmitida por un cromosoma X defectuoso.
Cuando decimos que la enfermedad de Fabry es una enfermedad de transmisión recesiva del cromosoma X, significa que, para que la enfermedad se manifieste en las mujeres, deben recibir los 2 cromosomas X defectuosos (uno del padre y otro de la madre).
Si un cromosoma X está sano y el otro está enfermo, la mujer no desarrollará la enfermedad, porque el cromosoma sano la protege de este defecto. Como los machos solo tienen un cromosoma X, no tienen forma de tener otro cromosoma X que los proteja. Por lo tanto, basta con un cromosoma X defectuoso para que desarrolle la enfermedad de Fabry.
Por lo explicado hasta ahora, es fácil entender por qué la enfermedad de Fabry es una afección que afecta mucho más a los hombres que a las mujeres. También es fácil entender por qué la enfermedad en los hombres se hereda siempre de la madre, ya que los varones humanos reciben necesariamente el cromosoma Y del padre y el cromosoma X de la madre.
Siguiendo este razonamiento, podemos afirmar que los hombres con la enfermedad de Fabry no pueden transmitirla a sus hijos varones, ya que solo reciben su cromosoma Y. Sin embargo, las hijas de padres con Fabry recibirán necesariamente un cromosoma X defectuoso. Si además reciben un cromosoma X defectuoso de su madre, desarrollarán la enfermedad.
En resumen:
- La enfermedad de Fabry prácticamente solo se da en los hombres.
- Para que una mujer tenga Fabry, necesita que ambos padres tengan el cromosoma X defectuoso*.
- Las mujeres suelen ser portadoras asintomáticas del cromosoma X defectuoso, porque su otro cromosoma X sano las protege de la enfermedad.
- Son las mujeres portadoras del cromosoma X defectuoso las que transmiten el defecto a sus hijos.
* Este concepto ha cambiado en los últimos años, ya que hay casos reconocidos de mujeres con un solo cromosoma X defectuoso que muestran diversos grados de manifestaciones clínicas de la enfermedad de Fabry.
Causas
Como se ha mencionado anteriormente, la enfermedad de Fabry es causada por una deficiencia de una enzima llamada alfa-galactosidasa A (alfa-gal A). Se trata de una enzima presente en el interior de nuestras células que se encarga de eliminar una sustancia grasa llamada globotriaosilceramida (GB3 o GL-3).
En ausencia de alfa-gal A, el GB3 se acumula dentro de las células y se deposita en varios tejidos, en particular en las paredes de los vasos sanguíneos. Por lo tanto, la enfermedad de Fabry forma parte de un grupo de trastornos llamados enfermedades de almacenamiento.
Todos los signos y síntomas de la enfermedad de Fabry se derivan de esta acumulación de GB3, que puede obstruir los vasos sanguíneos, causando isquemia e infartos de órganos y tejidos.
Síntomas
Los síntomas de la enfermedad de Fabry suelen seguir un determinado orden de aparición. Los primeros signos aparecen en la infancia o en la adolescencia temprana e incluyen:
Dolor en las extremidades
El dolor se produce principalmente en las manos y los pies. Está causada por la afectación de los nervios del sistema nervioso periférico. Suele ser el primer síntoma y puede desencadenarse por el estrés, el frío, el calor o la actividad física extenuante.
Lesiones de la piel
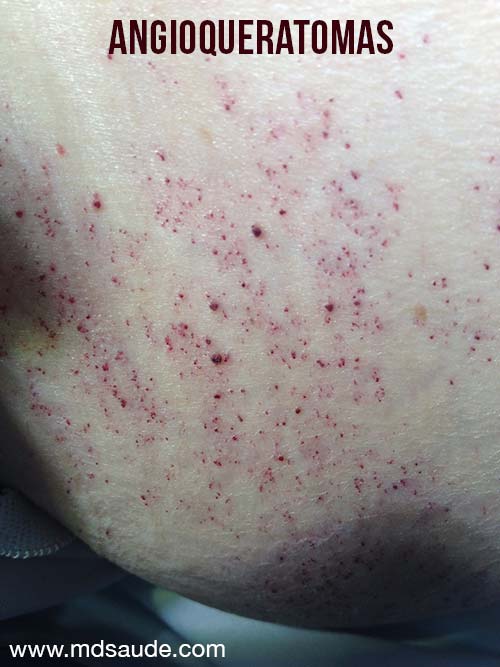
Dos lesiones son comunes: las telangiectasias y los angioqueratomas.
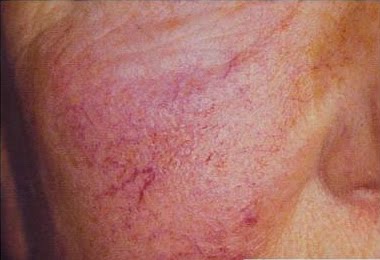
Las telangiectasias son pequeños vasos dilatados en la piel, también llamados arañas vasculares por su forma. Se observan con frecuencia en personas con venas varicosas en las piernas. Los angioqueratomas, en cambio, son elevaciones puntiformes de color púrpura.
En la enfermedad de Fabry, las dos lesiones suelen aparecer juntas, principalmente en los muslos, las caderas y alrededor del ombligo.
Cambios en las glándulas sudoríparas
Otro problema frecuente en Fabry es la afectación de las glándulas sudoríparas, responsables de la producción de sudor. Los pacientes pueden presentar una baja tasa de transpiración (hipohidrosis), temperatura corporal elevada, fatiga intensa y intolerancia al calor.
Cambios en los ojos
Los depósitos de GB3 en los vasos corneales de los ojos suelen causar la llamada verticilata corneal, una lesión de la córnea que puede detectarse con exámenes oculares especiales (oftalmoscopia con lámpara de hendidura).
Síntomas gastrointestinales
También son frecuentes los síntomas gastrointestinales, como el dolor después de comer, las náuseas y la diarrea.
Riñones
El 50 % de los pacientes con la enfermedad de Fabry presentan afectación renal. Los primeros signos son el aumento de la producción de orina debido a la incapacidad del riñón para retener agua. A esto le sigue la proteinuria y, posteriormente, la insuficiencia renal crónica. Una gran proporción de pacientes acaba necesitando hemodiálisis.
Corazón
La afectación cardíaca suele adoptar la forma de insuficiencia cardíaca, arritmias cardíacas, hipertrofia ventricular izquierda e isquemia miocárdica.
Cerebro
La afectación del sistema nervioso central puede adoptar la forma de accidente isquémico transitorio o derrame cerebral, mareos, vértigo o dolor de cabeza.
La enfermedad de Fabry en las mujeres
La mayoría de las mujeres son portadoras de un solo cromosoma X defectuoso y presentan pocos o ningún síntoma de la enfermedad de Fabry.
Sin embargo, debido a factores aún no bien conocidos, hay un pequeño grupo que desarrolla la enfermedad igual que los hombres, aunque sólo tenga un cromosoma defectuoso.
Diagnóstico
Dado que es una enfermedad rara y poco conocida, incluso entre los médicos, los pacientes suelen tener que esperar años entre los primeros síntomas y el diagnóstico definitivo. Cuando hay antecedentes familiares, el diagnóstico es más fácil porque la propia familia ya sugiere al médico la hipótesis de la enfermedad.
La tríada alteraciones renales + angioqueratomas + dolor en las extremidades en niños o varones jóvenes debe hacer sospechar siempre de la enfermedad de Fabry.
El diagnóstico puede confirmarse mediante la prueba de la enzima alfa-galactosidasa A. La mayoría de los pacientes tienen niveles indetectables o muy bajos.
Una biopsia de piel o de riñón puede proporcionar un diagnóstico en aquellos casos atípicos en los que el médico no considera inicialmente la enfermedad de Fabry como diagnóstico diferencial.
Las mujeres portadoras del gen de la enfermedad pueden ser asintomáticas y tener niveles normales de alfa-gal A. En estos casos, sólo las pruebas genéticas pueden confirmar si la mujer tiene el gen de la enfermedad y puede transmitirlo a sus hijos. El análisis genético se realiza todavía en muy pocos laboratorios.
Tratamiento
La enfermedad de Fabry no tiene cura, pero ya existen opciones terapéuticas eficaces capaces de reducir la acumulación de globotriaosilceramida (Gb3), retrasar la progresión de la enfermedad y mejorar la calidad de vida de los pacientes.
El tratamiento debe iniciarse lo antes posible, preferiblemente antes de la aparición de lesiones irreversibles, como fibrosis cardíaca o enfermedad renal avanzada.
Terapia de reemplazo enzimático (TRE)
El principal enfoque terapéutico en las últimas dos décadas ha sido la terapia de reemplazo enzimático (TRE), que consiste en la administración intravenosa de la enzima alfa-galactosidasa A recombinante. Actualmente, hay dos formulaciones disponibles:
- Agalsidasa alfa (Replagal®, Takeda): administrada cada dos semanas, por vía intravenosa, a la dosis de 0,2 mg/kg.
- Agalsidasa beta (Fabrazyme®, Sanofi Genzyme): también administrada quincenalmente, a la dosis de 1 mg/kg.
Los estudios demuestran que la TRE es capaz de reducir los depósitos de Gb3 en diversos tejidos, disminuir la inflamación vascular y estabilizar o retrasar la progresión de la disfunción renal, cardíaca y neurológica. Aun así, los resultados clínicos son variables, dependiendo del estadio de la enfermedad al inicio del tratamiento, del sexo del paciente y del fenotipo de la mutación genética.
A pesar de la reducción de la carga lisosomal y de la mejoría de algunos síntomas, el impacto de la TRE sobre la mortalidad a largo plazo todavía se está evaluando. Investigaciones observacionales, como el FOS (Fabry Outcome Survey) y el metarregistro europeo, indican beneficio en eventos clínicos importantes, especialmente cuando la terapia se inicia de forma precoz.
Chaperonas farmacológicas – Migalastat (Galafold®)
El migalastat (Galafold®, Amicus Therapeutics) es una terapia oral aprobada en Europa (desde 2016) y en EE. UU. (desde 2018), indicada para pacientes adultos con mutaciones genéticas específicas “susceptibles de respuesta” —que representan alrededor del 35 % al 50 % de los casos.
El migalastat es una chaperona farmacológica, es decir, una pequeña molécula que se une a ciertas formas inestables de la enzima alfa-galactosidasa A, estabilizando su estructura. Esto permite que la enzima llegue a los lisosomas y desempeñe su función de degradar el Gb3.
El migalastat se administra por vía oral en días alternos, lo que lo convierte en una opción atractiva para pacientes con un perfil genético favorable y que desean evitar la vía intravenosa. Estudios como el FACETS y el ATTRACT demostraron su eficacia en la estabilización de la función renal y cardíaca en pacientes con mutaciones responsivas.
Nuevas terapias en desarrollo
En los últimos años han surgido enfoques innovadores con potencial para revolucionar el tratamiento de la enfermedad de Fabry:
- Terapia génica: consiste en la introducción de una copia funcional del gen GLA en el organismo del paciente, con el objetivo de permitir la producción endógena y sostenida de la enzima alfa-galactosidasa A. Ensayos clínicos en fase 1/2 con vectores virales, como el FLT190 y el ST-920, han mostrado un aumento sostenido de la actividad enzimática en humanos.
- Terapia de reducción de sustrato (SRT): estrategia que busca inhibir la síntesis de glucoesfingolípidos, reduciendo la sobrecarga lisosomal. Nuevos inhibidores orales están en desarrollo, como el lucerastat y el venglustat, pero aún no están aprobados para uso clínico.
- Chaperonas alternativas y bifuncionales: además del migalastat, se están estudiando otras moléculas para aumentar la estabilidad de la enzima, incluso en combinaciones con TRE.
Pronóstico e impacto del tratamiento
La expectativa de vida de los pacientes no tratados se reduce de forma significativa, especialmente en los hombres con formas clásicas de la enfermedad, que pueden evolucionar a insuficiencia renal terminal, miocardiopatía grave y eventos cerebrovasculares precoces. En promedio, la supervivencia ronda los 50 años, pudiendo ser mayor en mujeres o en casos con fenotipos atenuados.
Con el tratamiento adecuado e iniciado de forma precoz, especialmente antes del compromiso irreversible de órganos diana, muchos pacientes pueden alcanzar una supervivencia cercana a la de la población general, con reducción de la progresión de la enfermedad y mejoría de la calidad de vida.
El tratamiento, ya sea con TRE o migalastat, es crónico y continuo, y exige seguimiento multidisciplinar con nefrólogo, cardiólogo, genetista, entre otros especialistas.
Referencias
- Fabry Disease – National Organization for Rare Disorders.
- Fabry disease – U.S National Library of Medicine.
- Fabry disease: Clinical features and diagnosis – UpToDate.
- Fabry disease: Treatment – UpToDate.
- Fabry Disease – Medscape.
- Long-term effectiveness of agalsidase alfa enzyme replacement in Fabry disease: A Fabry Outcome Survey analysis – Molecular genetics and metabolism reports.
- Migalastat: A Review in Fabry Disease – Drugs.
- Twenty years of the Fabry Outcome Survey (FOS): insights, achievements, and lessons learned from a global patient registry – Orphanet journal of rare diseases.



Dudas de los lectores sobre este tema
Preguntas reales enviadas por lectores y seleccionadas por el editor por su relevancia para este artículo.