¿Qué es la miastenia gravis?
La miastenia gravis (MG) es una enfermedad neuromuscular de origen autoinmune que, en su forma más común, provoca distintos grados de fatiga y debilidad muscular, afectando preferentemente los músculos de contracción voluntaria, como los de los brazos, piernas, rostro, ojos y los músculos torácicos responsables de la respiración.
La debilidad muscular asociada a la miastenia gravis suele empeorar tras la actividad física o cuando el paciente está más fatigado, como por ejemplo al final del día. Los síntomas tienden a mejorar con el descanso.
La miastenia es una enfermedad grave, pero que en las últimas décadas ha sido controlada adecuadamente gracias a nuevos esquemas de tratamiento, lo que ha permitido que la mayoría de los pacientes se mantenga activa y con buena calidad de vida.
En este artículo explicaremos qué causa la miastenia gravis, cuáles son sus síntomas, cómo se realiza su diagnóstico y qué tratamientos están actualmente disponibles.
Causas
La miastenia gravis es una enfermedad de origen autoinmune provocada por un defecto en la transmisión de los impulsos nerviosos hacia los músculos.
Una enfermedad autoinmune surge cuando nuestro sistema inmunitario comienza a producir autoanticuerpos de forma inapropiada, es decir, anticuerpos que atacan y destruyen elementos y estructuras sanas de nuestro propio organismo (si desea profundizar en el tema, lea el siguiente artículo: Enfermedad autoinmune).
En la miastenia gravis, el sistema inmunitario produce anticuerpos que dañan los receptores musculares encargados de recibir las señales enviadas por los nervios. Vamos a explicar cómo funciona la interacción entre los nervios y los músculos para que pueda entender mejor el origen de esta enfermedad.
¿Cómo provocan los autoanticuerpos la miastenia gravis?
Toda contracción muscular que usted realiza está controlada por impulsos nerviosos que se originan en el cerebro. Solo puede levantar el brazo, mover la cabeza o caminar, por ejemplo, porque su cerebro es capaz de enviar impulsos eléctricos que viajan por la médula espinal, alcanzan los nervios periféricos y llegan hasta las fibras musculares que necesitan ser activadas.
Una persona que sufre un traumatismo en la columna vertebral y presenta una lesión grave en la médula puede dejar de caminar porque las señales eléctricas que salen del cerebro ya no consiguen llegar a los músculos de las piernas.
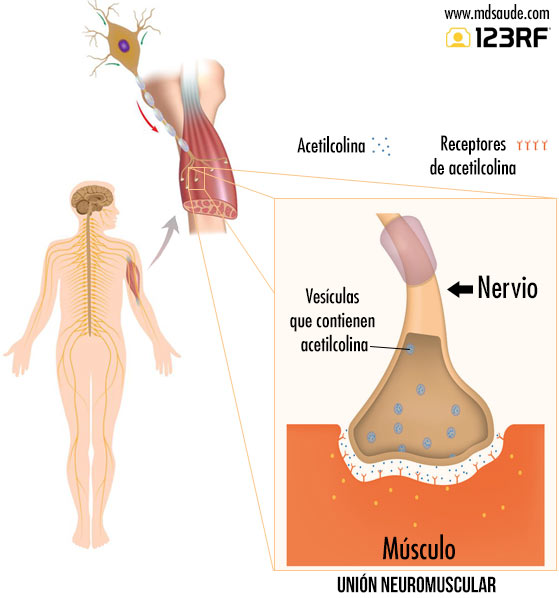
En la miastenia gravis, el problema no se encuentra en la médula espinal, sino al final del recorrido, cuando las fibras nerviosas se conectan con las fibras musculares. En realidad, los nervios no se conectan directamente con el músculo: existe un pequeño espacio entre la terminación nerviosa y la fibra muscular, conocido como unión neuromuscular. Consulte la ilustración adjunta para entenderlo mejor.
Cuando el impulso nervioso alcanza la terminación nerviosa, estimula la liberación de un neurotransmisor llamado acetilcolina. La acetilcolina se libera en la unión neuromuscular y es captada por los receptores de acetilcolina presentes en el músculo. Es esta captación de acetilcolina en la unión neuromuscular la que estimula la contracción muscular.
La miastenia gravis es una enfermedad que ocurre cuando, por razones aún desconocidas, el sistema inmunitario comienza a producir anticuerpos contra los receptores de acetilcolina presentes en los músculos. La acción de estos autoanticuerpos puede destruir o dañar hasta el 80 % de dichos receptores, lo que reduce drásticamente la capacidad de captar acetilcolina, especialmente durante el esfuerzo, momento en que el músculo necesita mayores cantidades de este neurotransmisor para funcionar correctamente.
¿Quiénes corren mayor riesgo?
La miastenia gravis es una enfermedad poco frecuente, que afecta aproximadamente entre 100 y 400 personas por cada millón de habitantes (un 0,01 a un 0,04 % de la población).
La miastenia gravis puede presentarse en ambos sexos, pero es más común en las mujeres: de cada 5 personas afectadas, 3 son mujeres. Además de ser más frecuente en el sexo femenino, la miastenia gravis también suele manifestarse más temprano en este grupo, generalmente en la tercera década de la vida. La edad media de inicio de los síntomas en las mujeres es de 28 años.
En los hombres, en cambio, la enfermedad es más común a partir de la quinta década de vida, siendo 42 años la edad media de aparición de los síntomas. Esta diferencia en la edad de inicio hace que, a partir de los 50 años, la miastenia gravis sea más frecuente en el sexo masculino.
Los recién nacidos hijos de madres con miastenia pueden presentar una forma transitoria de miastenia gravis llamada miastenia gravis neonatal, que aparece como resultado del paso de autoanticuerpos maternos a través de la placenta. Esta forma de MG dura pocas semanas, que es el tiempo que el organismo del bebé necesita para eliminar los anticuerpos maternos anómalos. Si se diagnostica y trata adecuadamente, más del 90 % de los bebés se curan sin secuelas. En raras ocasiones, la miastenia gravis en recién nacidos se debe a una enfermedad propia del neonato.
Varios estudios han sugerido una asociación entre la miastenia gravis y otras enfermedades autoinmunes, como la neuromielitis óptica, las enfermedades autoinmunes de la tiroides, el lupus eritematoso sistémico y la artritis reumatoide.
Síntomas
La característica principal de la miastenia gravis es un cuadro de fatiga muscular limitado a determinados grupos musculares, que puede ser fluctuante, es decir, con períodos de mejoría alternados con fases de empeoramiento. La fatiga muscular en la MG no se manifiesta como una sensación generalizada de cansancio, sino como una disminución aislada de la fuerza en ciertos músculos.
La debilidad muscular puede variar a lo largo del día, siendo habitualmente más intensa por la noche o después de algún esfuerzo físico. En las fases iniciales de la enfermedad, los síntomas pueden estar ausentes al despertar y aparecer a medida que avanza el día. A medida que la enfermedad progresa, los períodos libres de síntomas desaparecen. El paciente pasa a presentar debilidad en todo momento, que solo varía en intensidad a lo largo del día.
Aunque la miastenia puede provocar debilidad en cualquier grupo muscular de contracción voluntaria, existen algunas formas de presentación que son bastante características, como veremos a continuación.
Síntomas oculares
Más del 50 % de los pacientes presentan síntomas oculares al inicio de la enfermedad, siendo los más comunes la ptosis (caída del párpado) y la diplopía (visión doble). Entre los pacientes que comienzan con manifestaciones oculares, la mitad desarrollará enfermedad generalizada en un plazo de dos años.
En aproximadamente el 15 % de los casos, el paciente presenta únicamente síntomas oculares, sin progresar a una forma generalizada. Este cuadro se denomina miastenia gravis ocular.
Síntomas en la cara y el cuello
Alrededor del 15 % de los pacientes presentan síntomas relacionados con los músculos de la cara y el cuello, que incluyen: disartria (dificultad para articular palabras y frases), disfagia (dificultad para tragar), debilidad para masticar y pérdida de las expresiones faciales.
También es frecuente la debilidad en los músculos del cuello, lo que provoca dificultad para mantener la cabeza erguida.
Síntomas en las extremidades
La afectación de las extremidades es común, pero la debilidad aislada de una extremidad se presenta en menos del 5 % de los casos. Cuando las extremidades se ven afectadas, el paciente suele tener también debilidad en otros grupos musculares del cuerpo. La debilidad en los brazos y las manos es más común que en las piernas.
Síntomas respiratorios
La afectación de los músculos respiratorios produce los síntomas más graves de la miastenia gravis. La debilidad muscular respiratoria puede provocar insuficiencia respiratoria o incluso paro respiratorio, una situación grave conocida como crisis miasténica.
La crisis miasténica puede surgir espontáneamente durante las fases de agravamiento de los síntomas, o bien ser desencadenada por diversos factores, como cirugías, traumatismos, infecciones o medicamentos. Entre los fármacos que pueden empeorar la miastenia y aumentar el riesgo de una crisis miasténica, se encuentran:
- Antibióticos: aminoglucósidos (p. ej.: gentamicina o neomicina), quinolonas (p. ej.: ciprofloxacino, levofloxacino y norfloxacino), vancomicina, azitromicina y clindamicina.
- Anestésicos que provocan bloqueo muscular.
- Betabloqueantes (p. ej.: atenolol, labetalol, metoprolol y propranolol).
- Toxina botulínica (Botox).
- Cloroquina.
- Quinina.
- Procainamida.
- Magnesio.
Evolución natural de la enfermedad
En las fases iniciales de la enfermedad, los síntomas suelen ser transitorios, y el paciente puede permanecer horas, días o incluso semanas sin manifestaciones. Sin embargo, con el paso de los meses, las manifestaciones tienden a empeorar y se vuelven más persistentes. Aproximadamente el 80 % de los pacientes alcanzan el pico de la enfermedad en un plazo de 3 años.
La evolución de la miastenia gravis puede dividirse en tres fases:
- Durante los primeros 3 a 5 años de enfermedad, se presenta una fase activa con fluctuaciones y agravamiento progresivo de los síntomas. La mayoría de las crisis miasténicas ocurren en esta etapa. Más de la mitad de las muertes causadas por la miastenia tienen lugar en los primeros años de evolución.
- La segunda fase es una fase de estabilización. El paciente mantiene los síntomas, pero estos no se agravan. Las reagudizaciones suelen aparecer solo en el contexto de infecciones, uso de medicamentos que empeoran la debilidad muscular o cambios en la estrategia terapéutica de la miastenia, como una reducción en la dosis de los fármacos.
- Algunos pacientes progresan a la fase 3, en la cual puede producirse una remisión total de la enfermedad, permaneciendo el paciente libre de síntomas, con o sin necesidad de continuar con el tratamiento farmacológico.
Diagnóstico
El diagnóstico de la miastenia gravis se realiza mediante la exploración neurológica, que puede incluir pruebas como la prueba del hielo o la prueba con edrofónio, junto con exámenes complementarios como la electroneuromiografía.
Prueba del hielo
La prueba del hielo puede aplicarse en pacientes que presentan ptosis (caída del párpado). Se coloca una bolsa de hielo sobre el párpado afectado durante 2 minutos. El enfriamiento del músculo mejora la transmisión neuromuscular, permitiendo que el paciente abra el ojo temporalmente. Esta prueba tiene una efectividad aproximada del 80 %.
Prueba con edrofónio
El edrofónio es un fármaco con acción anticolinesterasa. Esto significa que inhibe la degradación de la acetilcolina presente en la unión neuromuscular, aumentando el tiempo que los escasos receptores de acetilcolina disponibles en el músculo tienen para captar el neurotransmisor. Este aumento de disponibilidad de acetilcolina ayuda al paciente a recuperar temporalmente la fuerza muscular. El edrofónio se administra por vía intravenosa, su efecto comienza en cuestión de segundos y dura entre 5 y 10 minutos.
La prueba del hielo suele preferirse frente a la del edrofónio debido al mayor riesgo de efectos adversos de este último, como desencadenar crisis asmáticas o eventos cardíacos.
Serología – Detección de autoanticuerpos
El diagnóstico serológico de la miastenia gravis se basa en la detección de autoanticuerpos circulantes dirigidos contra componentes de la unión neuromuscular. Los dos principales tipos son:
- Anticuerpos contra el receptor de acetilcolina (anti-AChR): son los más comunes, estando presentes en aproximadamente el 80–90 % de los pacientes con MG generalizada. En casos de miastenia ocular pura, la positividad se reduce al 40–50 %.
- Anticuerpos contra la tirosina quinasa muscular específica (anti-MuSK): menos frecuentes pero clínicamente relevantes, están presentes en hasta el 40–50 % de los pacientes con MG generalizada que son negativos para anti-AChR. En las formas oculares puras, estos anticuerpos suelen estar ausentes.
Además de los autoanticuerpos anti-AChR y anti-MuSK, un tercer tipo —el anti-LRP4— puede detectarse en una parte de los pacientes con miastenia gravis seronegativa. Está presente en hasta un 10 % de estos casos, especialmente en formas oculares o leves de la enfermedad.
Aunque aún no está disponible en todos los laboratorios, la inclusión del anti-LRP4 en paneles diagnósticos avanzados ha contribuido a confirmar el diagnóstico de MG en pacientes que carecen de los marcadores clásicos.
Aproximadamente un 5 % de los pacientes con miastenia gravis no presentan ninguno de estos autoanticuerpos detectables y se clasifican como seronegativos. En estos casos, el diagnóstico depende fundamentalmente de la evaluación clínica y de las pruebas electrofisiológicas. Los pacientes seronegativos tienden a presentar formas más leves de la enfermedad, muchas veces limitadas a los músculos oculares, aunque esto no constituye una regla absoluta.
Tratamiento
El tratamiento de la miastenia gravis se basa en una combinación de enfoques destinados tanto al alivio de los síntomas como al control del proceso autoinmune subyacente. La elección de la terapia depende de la gravedad de la enfermedad, del subtipo serológico, de la respuesta individual a los medicamentos y de la presencia de comorbilidades. Los principales tipos de tratamiento incluyen:
1. Inhibidores de la colinesterasa
Los inhibidores de la colinesterasa son medicamentos que aumentan la disponibilidad de acetilcolina en las uniones neuromusculares, mejorando la transmisión del impulso nervioso a los músculos y, en consecuencia, la fuerza muscular.
El principal representante de esta clase es la piridostigmina, que suele administrarse por vía oral y tiene un inicio de acción rápido. Aunque es eficaz para aliviar los síntomas en muchos pacientes, su respuesta es variable —en algunos casos, los efectos son discretos o insuficientes—.
Este tipo de medicación actúa únicamente sobre los síntomas y no modifica la evolución de la enfermedad ni interfiere en el proceso autoinmune que causa la miastenia.
Entre los efectos secundarios comunes se incluyen cólicos abdominales, diarrea, náuseas, salivación excesiva, sudoración aumentada y, en casos de sobredosis, crisis colinérgica, una condición caracterizada por exceso de acetilcolina en el organismo que puede provocar debilidad muscular intensa, sudoración excesiva, salivación, diarrea e incluso insuficiencia respiratoria.
2. Inmunosupresores
Dado que la miastenia gravis es una enfermedad autoinmune, los tratamientos más eficaces a largo plazo son aquellos que reducen o modulan la respuesta inmunitaria, disminuyendo la producción de autoanticuerpos que atacan los receptores de acetilcolina.
Los inmunosupresores pueden dividirse en dos categorías: convencionales y biológicos.
Inmunosupresores convencionales:
- Prednisona (corticoide): suele ser el primer inmunosupresor utilizado. A menudo se inicia con dosis altas que luego se reducen gradualmente.
- Azatioprina.
- Micofenolato mofetilo.
- Ciclosporina.
- Ciclofosfamida.
Estos fármacos son eficaces, pero actúan de forma generalizada sobre el sistema inmunitario, lo que puede aumentar el riesgo de infecciones, alteraciones hepáticas, hematológicas y otros efectos adversos sistémicos.
Inmunobiológicos (terapias dirigidas):
- Rituximab: anticuerpo monoclonal anti-CD20, eficaz especialmente en pacientes con anticuerpos anti-MuSK y formas refractarias de la enfermedad.
- Eculizumab: anticuerpo monoclonal que inhibe el componente C5 del sistema del complemento. Está indicado para pacientes con MG generalizada, positivos para anticuerpos anti-AChR y refractarios a los tratamientos convencionales. Ha demostrado beneficios significativos en ensayos clínicos, pero su coste es elevado y requiere vacunación previa contra el meningococo por el riesgo de infección grave.
- Otros agentes en estudio o de uso más reciente incluyen ravulizumab (de acción prolongada) y zilucoplan, un inhibidor subcutáneo del complemento C5.
La tendencia actual es hacia una terapia más personalizada, basada en el subtipo de autoanticuerpo presente (anti-AChR, anti-MuSK, anti-LRP4), la gravedad clínica y la respuesta al tratamiento.
3. Tratamiento de la crisis miasténica
La crisis miasténica es una emergencia médica caracterizada por debilidad muscular grave, especialmente de los músculos respiratorios, que puede conducir a insuficiencia respiratoria. En estos casos, se requiere hospitalización inmediata, a menudo con soporte ventilatorio.
Las dos principales terapias de acción rápida son:
- Plasmaféresis: elimina directamente los autoanticuerpos circulantes.
- Inmunoglobulina intravenosa (IVIg): modula rápidamente la respuesta inmunitaria de forma eficaz.
La elección entre una u otra depende de la gravedad de la crisis, las comorbilidades del paciente, la disponibilidad y la experiencia del centro hospitalario. Ambas opciones tienen efectos transitorios y se utilizan como tratamiento de rescate hasta que los inmunosupresores comienzan a hacer efecto.
4. Timectomía (extirpación del timo)
La timectomía, o cirugía para extirpar el timo, está indicada principalmente en dos situaciones:
- Presencia de timoma, un tumor benigno o maligno del timo, que se presenta en aproximadamente el 10–15 % de los pacientes con MG.
- Pacientes con miastenia gravis generalizada, positivos para anticuerpos anti-AChR, sin timoma, generalmente entre los 18 y 60 años.
Estudios recientes han demostrado que la timectomía puede reducir significativamente la gravedad de la enfermedad, la necesidad de inmunosupresores y la frecuencia de exacerbaciones, incluso en ausencia de timoma.
Aunque no todos los pacientes logran una remisión completa, la mejoría sintomática puede ser duradera. La eficacia de la timectomía tiende a ser mayor cuando se realiza en los primeros años tras el diagnóstico.
Pronóstico y calidad de vida
Con el tratamiento adecuado, la mayoría de los pacientes con miastenia gravis experimentan una mejora significativa de los síntomas y logra mantener una vida normal o casi normal. En muchos casos, es posible alcanzar una remisión parcial o completa, con reducción e incluso suspensión de la medicación.
No obstante, la enfermedad puede presentar fluctuaciones y períodos de empeoramiento, lo que requiere un seguimiento continuo con un neurólogo.
Referencias
- Clinical manifestations of myasthenia gravis – UpToDate.
- Diagnosis of myasthenia gravis – UpToDate.
- Pathogenesis of myasthenia gravis – UpToDate.
- Overview of the treatment of myasthenia gravis – UpToDate.
- Myasthenia Gravis – Review Article – The New England Journal of Medicine.
- Myasthenia Gravis – Medscape.
- Myasthenia Gravis Fact Sheet – National Institute of Neurological Disorders and Stroke
- Randomized Trial of Thymectomy in Myasthenia Gravis – The New England Journal of Medicine.
- Jameson JL, et al., eds. Myasthenia gravis and other diseases of the neuromuscular junction. In: Harrison’s Principles of Internal Medicine. 20th ed. New York, N.Y.: The McGraw-Hill Companies; 2018.
- Simon RP, et al. Motor disorders. In: Clinical Neurology. 10th ed. New York, N.Y.: McGraw-Hill Education; 2018.




Dudas de los lectores sobre este tema
Preguntas reales enviadas por lectores y seleccionadas por el editor por su relevancia para este artículo.