O que é a doença de Fabry?
A doença de Fabry, também chamada de doença de Anderson-Fabry, é uma patologia hereditária rara, transmitida de modo recessivo pelo cromossoma X, o cromossoma sexual feminino.
Estima-se que apenas 1 em cada 20 a 40 mil homens apresente a mutação responsável pelos sintomas da doença.
Os pacientes com doenças de Fabry apresentam uma deficiência de uma enzima chamada alfa-galactosidase A, o que acarreta problemas renais, cardíacos e do sistema nervoso.
Neste artigo explicaremos a doença de Fabry, abordando a sua transmissão genética, as suas causas, os sintomas e as opções de tratamento.
Genética
Todos os humanos possuem dois cromossomas sexuais. Os homens possuem um cromossoma sexual Y e um cromossoma sexual X (homens são XY). Já as mulheres possuem dois cromossomas sexuais X e nenhum cromossoma Y (mulheres são XX).
Cada um dos cromossomas sexuais que possuímos foi transmitido por um dos pais. Por exemplo, quando um casal tem um filho homem, isso significa que o menino recebeu o cromossoma Y do seu pai e o cromossoma X da mãe, formando, assim, o par XY. Quando o casal tem uma menina, ela recebe um cromossoma X do pai e um cromossoma X da mãe, formando o par XX.
Portanto, como a mãe só é capaz de passar o cromossoma X para os seus filhos, quem “decide” o sexo da criança é sempre o cromossoma de origem paterna. Se o pai passar um Y, será um menino; se ele passar o X, será uma menina.
A doença de Fabry é uma doença transmitida por um cromossoma X defeituoso.
Quando dizemos que o Fabry é uma doença transmitida de modo recessivo pelo cromossoma X, isso significa que, para a doença se manifestar nas mulheres, elas precisam receber os 2 cromossomas X defeituosos (um do pai e outro da mãe).
Se um cromossoma X for saudável e o outro doente, a mulher não desenvolve a doença, pois o cromossoma saudável a protege desse defeito. Como os homens só apresentam um cromossoma X, eles não têm como ter outro cromossoma X para protegê-lo. Portanto, basta um cromossoma X defeituoso para que ele desenvolva a doença de Fabry.
Para mais explicações sobre genética, leia: Introdução à genética: conceitos básicos.
Pelo que foi explicado até aqui, fica fácil entender por que a doença de Fabry é uma patologia que acomete muito mais os homens que as mulheres. Também é fácil entender por que a doença nos homens é sempre herdada da mãe, já que os humanos do sexo masculino obrigatoriamente recebem o cromossoma Y do pai e cromossoma X da mãe.
Seguindo esse raciocínio, podemos afirmar que os homens que possuem a doença de Fabry não são capazes de transmiti-la a seus filhos homens, dado que esses recebem somente o seu cromossoma Y. Porém, as filhas de pais com Fabry irão obrigatoriamente receber um cromossoma X defeituoso. Se elas também receberem um cromossoma X doente da mãe, desenvolverão a doença.
Resumindo:
- A doença de Fabry praticamente só ocorre em homens.
- Para uma mulher ter Fabry, ela precisa que ambos os pais tenham o cromossoma X defeituoso*.
- As mulheres costumam ser portadoras assintomáticas do cromossoma X defeituoso, pois o seu outro cromossoma X saudável a protege da doença.
- São as mulheres portadoras do cromossoma X defeituoso que transmitem o defeito para os seus filhos.
* Esse conceito tem mudado nos últimos anos, pois há casos reconhecidos de mulheres com apenas um cromossoma X defeituoso que apresentam graus distintos de manifestações clínicas da doença de Fabry.
Causas
Como já dito no início do texto, a doença de Fabry acontece por uma deficiência de uma enzima chamada alfa-galactosidase A (alfa-gal A). Essa é uma enzima presente dentro de nossas células que é responsável pela eliminação de uma substância gordurosa chamada globotriaosilceramida (GB3 ou GL-3).
Na ausência da alfa-gal A, a GB3 se acumula dentro das células, depositando-se em vários tecidos, principalmente nas paredes dos vasos sanguíneos. Por isso, a doença de Fabry faz parte de um grupo de patologias chamado de doenças de depósito.
Todos os sinais e sintomas da doença de Fabry são derivados desse acúmulo de GB3, que pode obstruir vasos sanguíneos, causando isquemias e infartos de órgãos e tecidos.
Sintomas
Os sintomas da doença de Fabry costumam seguir uma certa ordem de aparecimento. Os primeiros sinais surgem na infância ou início da adolescência e incluem:
Dores nos membros
A dor ocorre principalmente nas mãos e nos pés. É causada por acometimento dos nervos do sistema nervoso periférico. Normalmente, esse é o primeiro sintoma e pode ser desencadeado por estresse, frio, calor ou atividades físicas extenuantes.
Lesões de pele
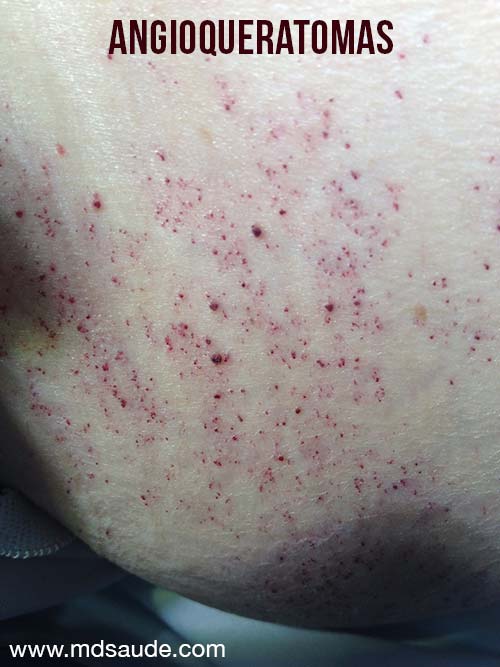
Duas lesões são comuns: teleangiectasias e angioqueratomas.
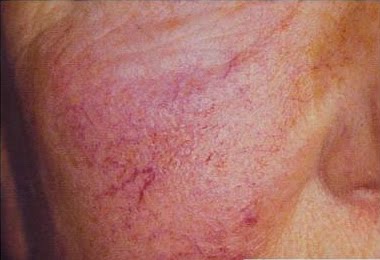
Teleangiectasias são pequenos vasos dilatados na pele, também chamados de aranhas vasculares pelo seu formato. São comumente vistos em pessoas com varizes nas pernas. Já os angioqueratomas são elevações puntiformes arroxeadas.
Na doença de Fabry, as duas lesões costumam aparecer juntas, principalmente nas coxas, quadril e em volta do umbigo.
Alterações das glândulas sudoríparas
Outro problema comum no Fabry é o acometimento das glândulas sudoríparas, responsáveis pela produção de suor. Os pacientes podem apresentar baixa taxa de transpiração (hipoidrose), elevada temperatura corporal, fadiga intensa e intolerância ao calor.
Alterações oculares
Depósitos de GB3 nos vasos da córnea dos olhos costumam causar a chamada córnea verticilata, uma lesão da córnea que pode ser detectada com exames oftalmológicos especiais (oftalmoscopia de lâmpada de fenda).
Sintomas gastrointestinais
Sintomas gastrointestinais como dor após a alimentação, náuseas e diarreia também são comuns.
Rins
50% dos pacientes com doença de Fabry apresentam acometimento renal. Os primeiros sinais são o aumento da produção de urina por incapacidade do rim em reter água. Depois surge a proteinúria e, mais adiante, a insuficiência renal crônica. Boa parte dos pacientes acaba por necessitar de hemodiálise.
Coração
O envolvimento cardíaco costuma se dar na forma de insuficiência cardíaca, arritmias cardíacas, hipertrofia do ventrículo esquerdo e isquemia do miocárdio.
Cérebro
O acometimento do sistema nervoso central pode se dar através de ataques isquêmicos transitórios ou AVC, tonturas, vertigens ou dor de cabeça.
Doença de Fabry nas mulheres
A maioria das mulheres é portadora de apenas um cromossoma X defeituoso e apresenta poucos ou nenhum sintoma da doença de Fabry.
Porém, por fatores ainda não bem conhecidos, existe um pequeno grupo que desenvolve a doença tal como os homens.
Diagnóstico
Por ser uma doença rara e pouco conhecida, inclusive entre os médicos, muitas vezes os pacientes precisam esperar anos entre os primeiros sintomas e o diagnóstico definitivo. Quando há história familiar, o diagnóstico sai mais facilmente, pois a própria família já sugere a hipótese da doença ao médico.
A tríade de alterações renais + angioqueratomas + dor nos membros em crianças ou jovens do sexo masculino, deve sempre levantar a suspeita de doença de Fabry.
O diagnóstico pode ser confirmado através da dosagem da enzima alfa-galactosidase A. A maioria dos pacientes apresenta níveis indetectáveis ou muito reduzidos.
A biópsia de pele ou do rim pode fornecer o diagnóstico naqueles casos atípicos, nos quais o médico não pensa inicialmente na doença de Fabry como diagnóstico diferencial.
As mulheres portadoras do gene da doença podem ser assintomáticas e ter níveis de alfa-gal A normais. Nestes casos, somente testes genéticos podem confirmar se a mesma possui o gene da doença, sendo passível de transmissão para seus filhos. A análise genética ainda é feita em pouquíssimos laboratórios.
Tratamento
A doença de Fabry não tem cura, mas já existem opções terapêuticas eficazes capazes de reduzir o acúmulo de globotriaosilceramida (Gb3), retardar a progressão da doença e melhorar a qualidade de vida dos pacientes.
O tratamento deve ser iniciado o mais precocemente possível, preferencialmente antes do surgimento de lesões irreversíveis, como fibrose cardíaca ou doença renal avançada.
Terapia de reposição enzimática (TRE)
A principal abordagem terapêutica nas últimas duas décadas tem sido a terapia de reposição enzimática (TRE), que consiste na administração intravenosa da enzima alfa-galactosidase A recombinante. Atualmente, duas formulações estão disponíveis:
- Agalsidase alfa (Replagal®, Takeda): administrada a cada duas semanas, por via intravenosa, na dose de 0,2 mg/kg.
- Agalsidase beta (Fabrazyme®, Sanofi Genzyme): também administrada quinzenalmente, na dose de 1 mg/kg.
Estudos demonstram que a TRE é capaz de reduzir os depósitos de Gb3 em diversos tecidos, diminuir a inflamação vascular e estabilizar ou retardar a progressão da disfunção renal, cardíaca e neurológica. Ainda assim, os resultados clínicos são variáveis, dependendo do estágio da doença no início do tratamento, do sexo do paciente e do fenótipo da mutação genética.
Apesar da redução da carga lisossomal e melhora de alguns sintomas, o impacto da TRE sobre a mortalidade a longo prazo ainda está sendo avaliado. Pesquisas observacionais, como o FOS (Fabry Outcome Survey) e o meta-registro europeu, indicam benefício em eventos clínicos importantes, especialmente quando a terapia é iniciada precocemente.
Chaperonas farmacológicas – Migalastate (Galafold®)
O migalastate (Galafold®, Amicus Therapeutics) é uma terapia oral aprovada na Europa (desde 2016) e nos EUA (desde 2018), indicada para pacientes adultos com mutações genéticas específicas “passíveis de resposta” — que representam cerca de 35% a 50% dos casos.
O migalastate é uma chaperona farmacológica — ou seja, uma pequena molécula que se liga a certas formas instáveis da enzima alfa-galactosidase A, estabilizando sua estrutura. Isso permite que a enzima chegue aos lisossomos e desempenhe sua função de quebrar o Gb3.
O migalastate é administrado por via oral, em dias alternados, o que o torna uma opção atrativa para pacientes com perfil genético favorável e que desejam evitar a via endovenosa. Estudos como o FACETS e o ATTRACT demonstraram sua eficácia na estabilização da função renal e cardíaca em pacientes com mutações responsivas.
Novas terapias em desenvolvimento
Nos últimos anos, têm surgido abordagens inovadoras com potencial de revolucionar o tratamento da doença de Fabry:
- Terapia gênica: consiste na introdução de uma cópia funcional do gene GLA no organismo do paciente, com o objetivo de permitir a produção endógena e sustentada da enzima alfa-galactosidase A. Ensaios clínicos em fase 1/2 com vetores virais, como o FLT190 e o ST-920, têm demonstrado aumento sustentado da atividade enzimática em humanos.
- Terapia de redução de substrato (SRT): estratégia que busca inibir a síntese de glicoesfingolipídios, reduzindo a sobrecarga lisossomal. Novos inibidores orais estão em desenvolvimento, como o lucerastat e o venglustat, mas ainda não estão aprovados para uso clínico.
- Chaperonas alternativas e bifuncionais: além do migalastate, outras moléculas estão sendo estudadas para aumentar a estabilidade da enzima, inclusive em combinações com TRE.
Prognóstico e impacto do tratamento
A expectativa de vida dos pacientes não tratados é significativamente reduzida, especialmente nos homens com formas clássicas da doença, que podem evoluir para insuficiência renal terminal, miocardiopatia grave e eventos cerebrovasculares precoces. Em média, a sobrevida gira em torno dos 50 anos, podendo ser maior em mulheres ou em casos com fenótipos atenuados.
Com o tratamento adequado e iniciado precocemente, especialmente antes do comprometimento irreversível de órgãos-alvo, muitos pacientes podem alcançar uma sobrevida próxima à da população geral, com redução da progressão da doença e melhora da qualidade de vida.
O tratamento, seja com TRE ou migalastate, é crônico e contínuo, exigindo seguimento multidisciplinar com nefrologista, cardiologista, geneticista, entre outros especialistas.
Referências
- Fabry Disease – National Organization for Rare Disorders.
- Fabry disease – U.S National Library of Medicine.
- Fabry disease: Clinical features and diagnosis – UpToDate.
- Fabry disease: Treatment – UpToDate.
- Fabry Disease – Medscape.
- Long-term effectiveness of agalsidase alfa enzyme replacement in Fabry disease: A Fabry Outcome Survey analysis – Molecular genetics and metabolism reports.
- Migalastat: A Review in Fabry Disease – Drugs.
- Twenty years of the Fabry Outcome Survey (FOS): insights, achievements, and lessons learned from a global patient registry – Orphanet journal of rare diseases.

Dúvidas de leitores sobre este tema
Perguntas enviadas por leitores e selecionadas pelo editor por sua relevância para este artigo.
Mais comentários dos leitores
Ótimo texto mais vi uma informação e fiquei preocupada tem tem doença de fabry só dura 50 anos
bom dia Dr. meu filho tem 28 anos e essa semana descobriu essa doença fabry, descobriu depois de um ecocardiograma com contraste de microbolhas, fez pque sentia dores no peito e o coraçao sempre acelerava, fazendo eletrocardiogramas investigando o problema cardiaco e pressao alta ( pressao sempre foi normal ) agora a pressao so fica alta. quero saber se corre risco de ter morte subita, ou corre algum outro risco . obrigada.
Boa matéria, só discordo num ponto, não é necessário que para uma mulher ter Fabry, o pai e mãe sejam portadores, somente 1. Minha filha é portadora da doença, pois herdou de meu esposo, que herdou da mãe dele…